
| 09:00-14:30 | Registration |
| 14:30-14:40 | Conference opening, T. Frederiksen |
| 14:40-16:20 | Mo1: Surface scattering and chemistry |
| 16:20-17:00 | Coffee break |
| 17:00-18:40 | Mo2: Solid-liquid interfaces |
| 19:00-21:00 | Welcome reception, Sala de Musica, Palacio Miramar |
Chair: U. Höfer, Marburg, Germany
Contributed talk
Role of the vibrational energy to enhance the dissociative adsorption of N2 on metal surfaces
1Donostia International Physics Center, 20018 San Sebastián, Spain
2Univ. Bordeaux and CNRS, ISM, UMR5255, F-33400, Talence, France
3Centro de Física de Materiales (CSIC-UPV/EHU), 20018 San Sebastián, Spain
4Dep. Física de Materiales, Apartado 1072, 20080 San Sebastián, Spain
5Humboldt Universität zu Berlin, Institut für Chemie, Unter den Linden 6, D-10009 Berlin, Germany
The dissociation of N2 on metal surfaces is usually the rate limiting step in the synthesis of many important compounds (ammonia, nitric acid, organic nitrates...) that are produced in chemical industry. In this theoretical study we investigate the efficiency of the vibrational energy to increase the dissociative adsorption of N2 on the Fe(110) and the W(110) surfaces. As shown in Refs. [1] and [2] for the non-vibrationally excited N2, dissociation, which is activated in both cases, is dominated by the energy barriers that appear when the molecule is close to the surface in the former [Fe(110)] and far from it in the latter [W(110)].
Here, we perform multidimensional molecular dynamics simulations on precalculated ab-initio potential energy surfaces to calculate the dissociative sticking probability as a function of the initial translational and vibrational energy of the molecule. Based on low dimensional schemes (see Fig.1), it has been thought that the vibrational energy would be efficient to promote dissociation in late barrier systems such as N2/Fe(110), but inefficient in early barrier systems such as N2/W(110). Our results show that though the vibrational energy is more efficient in the former, it is still efficient in the early barrier N2/W(110) system. The reason is that the vibrational energy not only allows to overcoming late barriers, but also can open new dissociation paths with lower early barriers than those found for N2 in the vibrational ground state. In particular, we observe that the new dissociation paths on the N2/W(110) system are actually dominated by late barriers. Therefore, there is no contradiction between our results and the original Polanyi's rules developed for gas-phase collisions [3].
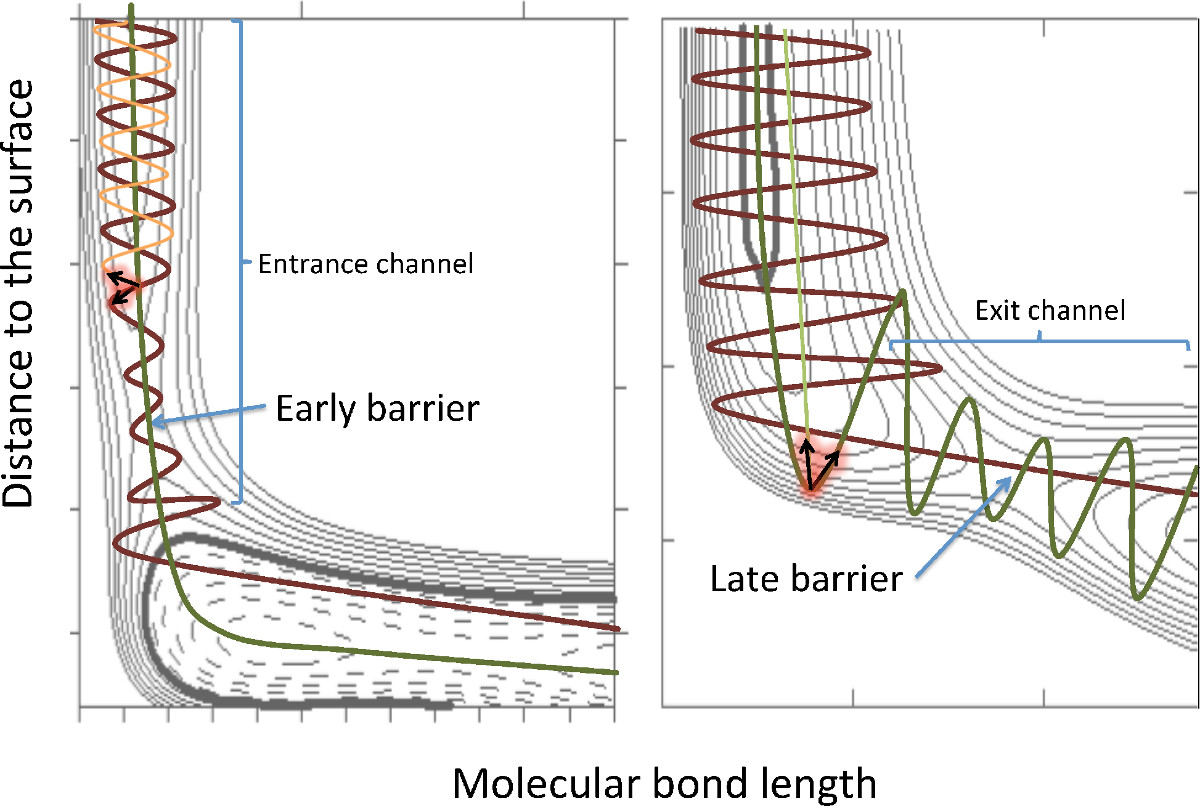
Figure 1: Examples of 2D cuts of early barrier and late barrier systems. The brown (green) line corresponds to the dissociating process of a molecule with (without) vibrational energy in the 2D simplified model.
[1] G. A. Bocan, R. Díez Muiño, M. Alducin, H. F. Busnengo, and A. Salin, J. Chem. Phys. 128, 154704 (2008)
[2] I. Goikoetxea, M. Alducin, R. Díez Muiño, and J. I. Juaristi, Phys. Chem. Chem. Phys. 14, 7471 (2012)
[3] J. C. Polanyi and W. H. Wong, J. Chem. Phys. 51, 1439 (1969)