
| 09:00-10:40 | Th1: Adsorbate and interface dynamics |
| 10:40-11:20 | Coffee break |
| 11:20-13:00 | Th2: STM-IETS and beyond |
| 13:00-15:30 | Lunch break (on your own) |
| 15:30-16:40 | Th3: Molecular films and 2D materials |
| 16:40-17:20 | Coffee break |
| 17:20-19:00 | Th4: Tip-enhanced vibrational spectroscopies |
| 20:30-23:00 | Conference dinner at Cofradía Vasca de Gastronomía, Old Town |
Chair: M. Alducin, San Sebastián, Spain
Contributed talk
Electronic and vibrational states of single Tin-Phthalocyanine molecules – a numerical STM study
1Institut für Physik, Technische Universität Ilmenau, Germany
2Key Laboratory for the Physics and Chemistry of Nanodevices, Peking University, China
3School of Chemistry, Newcastle University, United Kingdom
4Institut für Experimentelle und Angewandte Physik, Christian-Albrechts-Universität zu Kiel, Germany
Tin-Phthalocyanine (SnPc) molecules adsorb in two different stable configurations, the protruding central tin atom pointing towards (SnPc↓) or away from (SnPc↑) the surface. Electronic and vibrational properties of both configurations adsorbed on an ultrathin SnPc buffer film on Ag(111) have been investigated with scanning tunneling microscopy (STM) and density functional calculations, using a numerically very efficient approach based on the Tersoff-Hamann approximation. The characteristic features in spectra of the differential conductance are reproduced by the calculations including the remarkable difference between the two configurations. The first-principles approach going beyond the quasistatic approximation reveals that excitations of various vibrational quanta induce replica of orbital spectroscopic signatures for the configuration with a lower molecule–surface coupling (SnPc↑). Spectra of molecules with a larger coupling to the surface (SnPc↓) can be described by considering elastic tunneling to orbital resonances alone. The applicability of our perturbative approach is discussed regarding a possible breakdown of the Born-Oppenheimer approximation.
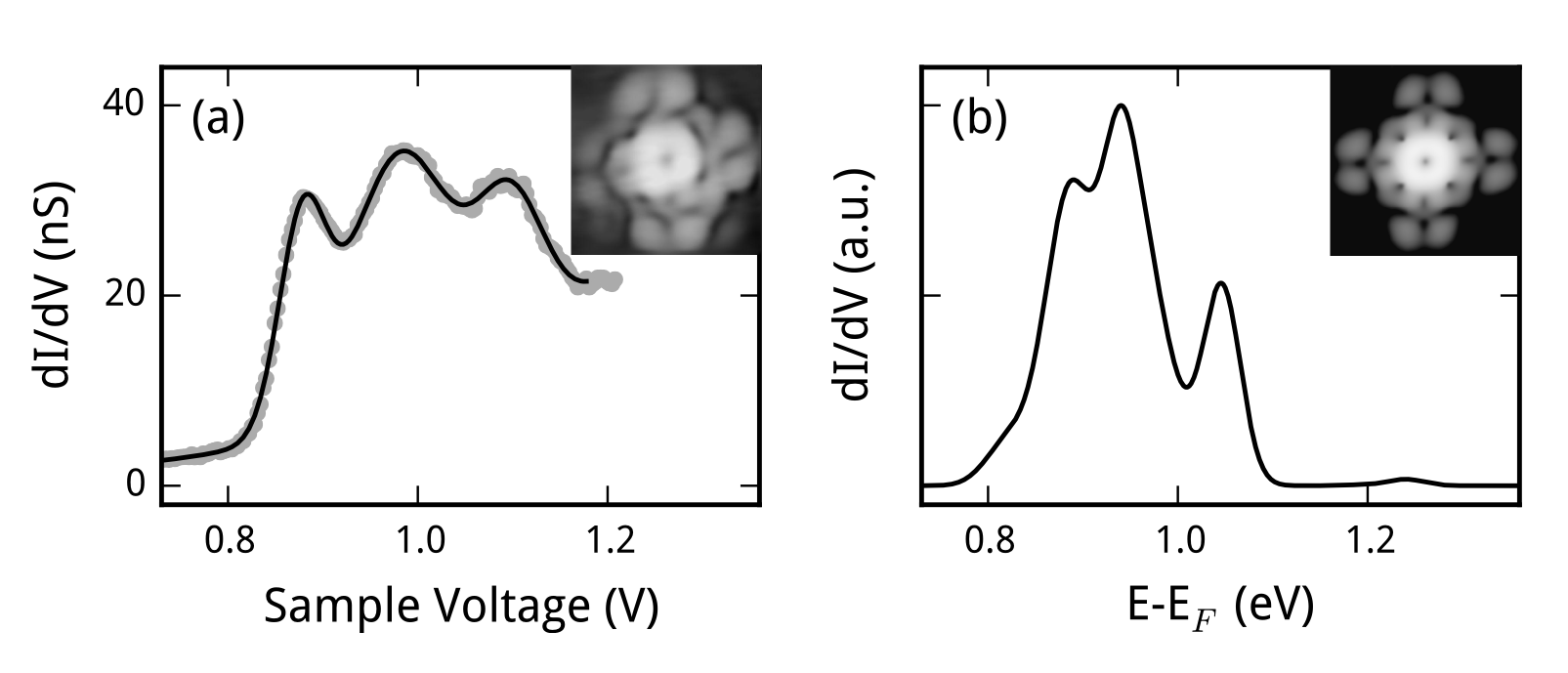
Figure 1: (a) Measured differential conductance (dI/dV) spectrum of SnPc↑ on a SnPc↓ buffer-layer on Ag(111) (gray dots). The full line represents a fit of three Gaussians and an exponential background to the experimental data. The spectrum shows three distinct peaks at 0.88, 0.98 and 1.10 eV. The spectra have been measured atop the center of the molecule. The inset shows the measured constant-current STM image. (b) Calculated dI/dV spectrum for the same configuration including contributions from vibrational excitations due to inelastic tunneling. In agreement with experiment, three peaks are present at 0.88, 0.93 and 1.04 eV, each of them comprising the spectroscopic signature of several molecular vibrational quanta. The purely elastic calculations fail to reproduce the three-peak structure (not shown). The inset shows the calculated constant-current STM image of SnPc↑.