
Invited talk
Imaging molecular motions with scanning tunnelling microscopy
1Faculty of Physics and Center for Nanointegration Duisburg-Essen (CeNIDE), University of Duisburg-Essen, Lotharstr. 1, D47057 Duisburg, Germany
2Centro de Física de Materiales (CSIC) and Donostia International Physics Center, Paseo de Manuel Lardizabal 5, E20018 San Sebastián-Donostia, Spain
3Institut de Sciences Moléculaires d'Orsay (CNRS), and Université Paris-Sud, UMR 8214, Bât. 351, F91405 Orsay Cédex, France
The scanning tunneling microscopy (STM) has a time resolution longer than typical molecular motion times. However, the molecular dynamics induced by the tunnelling electrons can nevertheless be probed by the STM. When it involves continuous transitions between several conformations, the real time characterization of the telegraph noise in the tunnelling current is a powerful tool to study STM-induced dynamics. We studied the case of individual copper phthalocyanine molecules on Cu(111) surfaces. The hopping rate between potential minima, the noise amplitude and the relative occupation of the involved states can be measured as a function of the tunnelling parameters, providing spatially resolved maps [1]. In contrast to standard STM, this technique gives access to transiently populated states revealing an electron-driven hindered rotation between the equilibrium and two metastablepositions of an individually adsorbed molecule [1,2]. A DFT based description of the adsorbed molecule reveals the adsorption properties of the molecule with a metastable state corresponding to a rotation of 7° of the molecule from equilibrium. A sudden approximation approach then allows the ab-initio treatment of the observed electron-induced transitions.
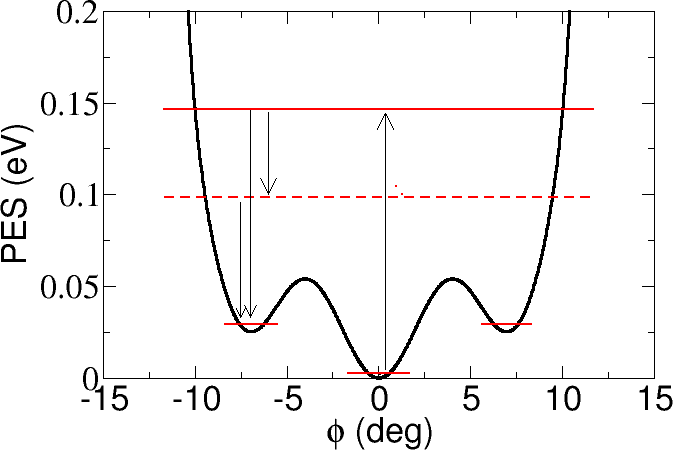
Figure 1: Potential energy surface of a copper phthalocyanine molecule with respect to the rotational angle of the molecule with respect to the Cu(111) surface. The molecule has two local and one global potential minima, and electrons can induce fluctuations among the three wells.
[1] J. Schaffert et al., Nat. Mat. 12, 223 (2013); Rev. of Scientific Instruments 84, 043702 (2013)
[2] J. Schaffert et al., Phys. Rev. B 88, 075410 (2013)