Poster
Femtosecond laser driven molecular dynamics on surfaces: O2 on Ag(110)
1Centro de Física de Materiales CFM/MPC (CSIC-UPV/EHU), San Sebastián, Spain
2Donostia International Physics Center (DIPC), San Sebastián, Spain
3Institut für Chemie, Universität Potsdam, Potsdam, Germany
4Departamento de Física de Materiales, Facultad de Químicas, San Sebastián, Spain
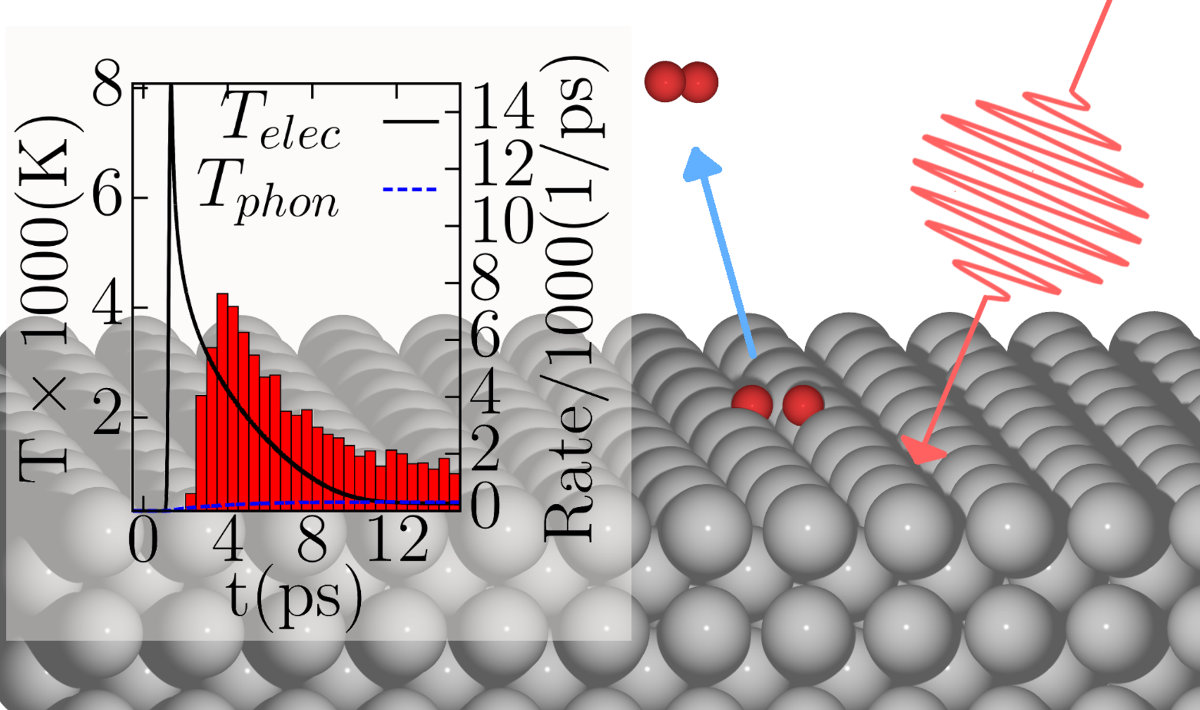
We present a model for simulating the six dimensional dynamics of an adsorbed molecule on a metal surface which is driven by a ultrashort (~100 fs) laser pulse. Both electron and phonon mediated processes are considered. As an example system we use O2 on the Ag(110) surface. This system is interesting because it features several adsorption wells, which can then show different dynamical behaviors depending on the three possible outcomes of the simulations: desorption of the molecule, dissociation of the molecule or molecule staying adsorbed in the adsorption well. We simulate the dynamics of the molecule by a classical Langevin model. The interaction of the surface and the molecule is described by a six dimensional potential energy surface which has been recently developed on the basis of semi-local density functional theory [1]. The effect of the laser pulse on the metal surface is described by the two temperature model (2TM), which consists in solving two coupled differential equations for the electron and phonon temperatures. Laser heating of the surface and phonon mediated processes enter the dynamics by setting the surface temperature at every time step in the Generalized Langevin Oscillator model (GLO) model to the phonon temperature calculated from the 2TM. The action of laser heated surface electrons on the molecule is modeled by friction and random forces exerted on the molecule. These random and frictional forces are linked via the fluctuation-dissipation theorem, and the variance of the random forces is proportional to the electronic temperature obtained from the 2TM. The corresponding friction coefficient is calculated in the basis of the local density friction approximation [2]. In fact, the model is similar to the one used in Ref. [3], but extended to incorporate phonons within the GLO. The dynamics calculations show that for O2 on Ag(110) inclusion of phonons is important, and that depending on the electronic density in which the molecule is embedded, it can significantly either enhance or reduce desorption probabilities.
Figure 1: Schematic illustration of the studied problem. In the inset we show electron and phonon temperatures of the surface and the desorption rate.
[1] I. Lončarić et al., Phys. Chem. Chem. Phys. (2015)
[2] J. I. Juaristi et al., Phys. Rev. Lett. 100, 116102 (2008)
[3] G. Füchsel et al., Phys. Chem. Chem. Phys. 13, 8659 (2011)