Poster
Theorical study of NO adsorption on Cu(110) and O(2×1)/Cu(110) surfaces
1DIPC (Donostia International Physics Center), Paseo Manuel de Lardizabal 4, Donostia-San Sebastián, E-20018, Spain
2Departamento de Física de Materiales, UPV/EHU, Facultad de Química, Donostia-San Sebastián, E-20080, Spain
3Centro de Física de Materiales (CFM-MPC), Centro Mixto CSIC-UPV/EHU, Paseo Manuel de Lardizabala 5, Donostia-San Sebastián, E-20018, Spain
4IKERBASQUE, Basque Foundation for Science, Bilbao, E-48011, Spain
Previous work showed the preferred tilted adsorption of Carbon monoxide (CO) on O(2×1)/Cu(110) surface [1]. As a consequence of the tilting, the dipole attraction between CO molecules resulted in the formation of one-dimensional molecular rows along the [110] direction. Recent scanning tunneling microscope (STM) experiments on nitric oxide (NO) adsorption on Cu(110) reveal also two possible orientations, vertical or tilted, depending on temperature [2].
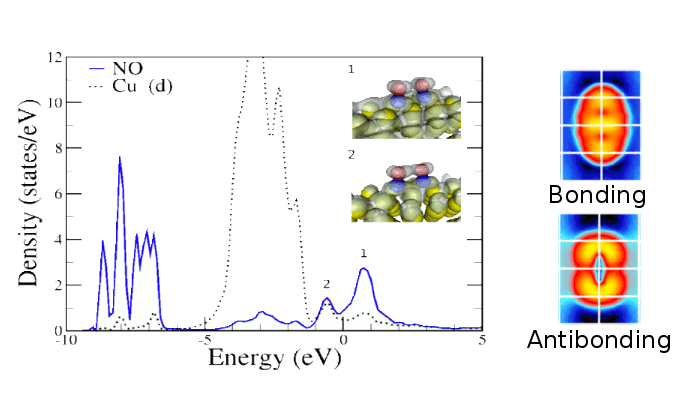
In this work we studied the interaction between NO molecules on Cu(110) and O(2×1)/Cu(110) using Density Functional Theory (DFT). Our results shows that NO interacts strongly with the Cu(110) surface, losing its spin polarization at all coverages. The dimer configuration is favored on a confronted tilted geometry (figure 1) along the [110]. On O(2×1)/Cu(110) surface NO adsorbs between CuO rows, in a way that N atoms produce a spontaneous pulling of the row Cu atoms. In this case, vertical and tilted adsorption along the [001] direction are energetically degenerate and the NO molecule show a spin polarization of 1/2 μB. In spite of the low energetic cost to tilt the molecule, we do not find any evidence that the formation of dimers or molecular rows are favored on the O(2×1)/Cu(110) surface [3].
Figure 1: PDOS representation. NO dimer on Cu(110) along the [110] direction. Interaction between 2π* molecular orbitals around the Fermi level.
[1] M. Feng, P. Cabrera-Sanfelix, C. Lin, A. Arnau, D. Sánchez-Portal, J. Zhao, P. M. Echenique and H. Petek, ACS Nano. 5, 8877-8883 (2011)
[2] A. Shiotari, Y. Kitaguchi, H. Okuyama, S. Hatta and T. Aruga, Phys. Rev. Lett. 106, 156104 (2011)
[3] A. X. Brión-Ríos, D. Sánchez-Portal, P. Cabrera-Sanfelix, Phys. Chem. Chem. Phys., submitted